When the skin speaks: Cutaneous manifestations as the first sign of hemophagocytic syndrome of unknown origin.
Dermatol Rev Mex. 2025; 69 (4): 603-606. https://doi.org/10.24245/dermatolrevmex.v69i4.10642
Neivy Izabal Tinajero,1 Eduardo González López,2 Estefanía Zepeda Muñoz,3 Pamela Palacios Vega3
1 Departamento de Pediatría.
2 Departamento de Hematología.
Hospital General Regional 1, Instituto Mexicano del Seguro Social, Baja California, México.
3 Departamento de Dermatología, Hospital Infantil de México Federico Gómez, Ciudad de México.
Estimado editor:
El síndrome hemofagocítico, también llamado linfohistiocitosis hemofagocítica, es una enfermedad rara y de alta mortalidad, con frecuencia infradiagnosticada debido a su manifestación clínica variable. Este síndrome se distingue por una respuesta inmunitaria exagerada mediada por linfocitos T citotóxicos y células natural killer desreguladas, lo que provoca inflamación sistémica, inestabilidad hemodinámica, insuficiencia multiorgánica y, en última instancia, la muerte del paciente. Los factores desencadenantes de este síndrome son diversos e incluyen: variantes patogénicas genéticas, infecciones, procesos oncológicos o reumatológicos; sin embargo, en algunos casos, se desconoce la causa de esta enfermedad. Es decisivo mantener un alto índice de sospecha para establecer el diagnóstico temprano y mejorar el pronóstico del paciente mediante el tratamiento oportuno.1,2,3
CASO CLÍNICO
Paciente femenina de 8 años, previamente sana, sin antecedentes familiares ni patológicos de importancia para el padecimiento actual. Tenía una dermatosis diseminada a la cabeza, el tronco y las extremidades superiores, que afectaba ambas mejillas, el tórax anterior y los brazos, caracterizada por máculas eritematosas, confluentes, mal delimitadas e irregulares, de dos semanas de evolución; asociada con dolor abdominal difuso y fiebre. Recibió tratamiento con mútliples antibióticos (lincosamidas, sulfonamidas y penicilinas) y tratamiento sintomático con antiespasmódicos y analgésicos, sin mejoría clínica.
A su ingreso, persistía la dermatosis y el dolor abdominal, tenía hepatomegalia y esplenomegalia. Se inició tratamiento con corticosteroide tópico de potencia media y emolientes; sin embargo, la dermatosis evolucionó con aparición de placas constituidas por vesículas y ampollas de diversos tamaños, de contenido seroso, además de costras hemáticas y algunas melicéricas, de bordes bien definidos e irregulares. Figura 1
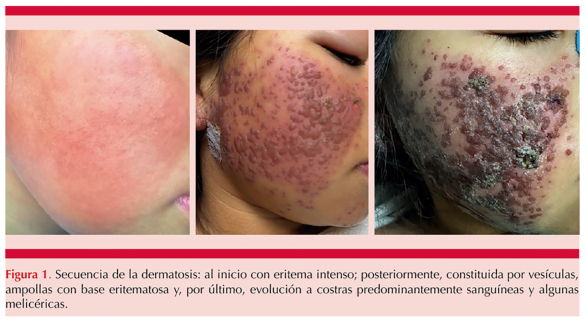
Tenía fiebre de alto grado y de difícil control e inestabilidad hemodinámica que ameritó su ingreso a la unidad de cuidados intensivos.
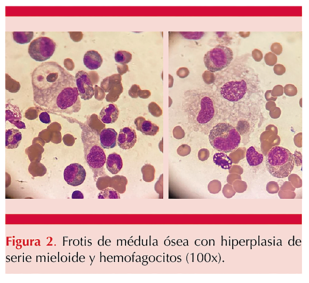
Se inició con el proceso diagnóstico infeccioso: policultivos, panel reumatológico y serologías para toxoplasmosis, rubéola, citomegalovirus, herpes simple, VIH, parvovirus y virus de Epstein-Barr, que resultaron negativos. Los resultados de laboratorio evidenciaron anemia, trombocitopenia y ferritina elevada. Con esto y la asociación con fiebre y hepatoesplenomegalia se integró la sospecha diagnóstica de síndrome hemofagocítico, por lo que se tomó biopsia de médula ósea, cuyo estudio reveló hiperplasia de la serie mieloide y hemofagocitosis. Figura 2
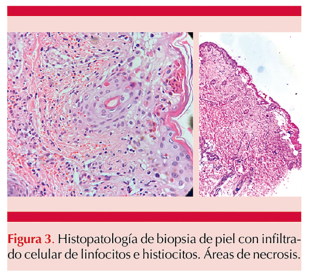
 El estudio de la biopsia cutánea mostró infiltrado linfohistiocítico perivascular con áreas de necrosis (Figura 3). Estos hallazgos confirmaron la sospecha diagnóstica. Se inició tratamiento conforme al protocolo HLH-04 con dexametasona, ciclosporina y etopósido. Además, se agregó rituximab y ruxolitinib al tratamiento por evolución tórpida. Sin embargo, a pesar del tratamiento instaurado, la paciente desarrolló síndrome de falla orgánica múltiple con desenlace fatal a los 36 días de estancia hospitalaria.
El estudio de la biopsia cutánea mostró infiltrado linfohistiocítico perivascular con áreas de necrosis (Figura 3). Estos hallazgos confirmaron la sospecha diagnóstica. Se inició tratamiento conforme al protocolo HLH-04 con dexametasona, ciclosporina y etopósido. Además, se agregó rituximab y ruxolitinib al tratamiento por evolución tórpida. Sin embargo, a pesar del tratamiento instaurado, la paciente desarrolló síndrome de falla orgánica múltiple con desenlace fatal a los 36 días de estancia hospitalaria.
DISCUSIÓN
El síndrome hemofagocítico es una enfermedad poco frecuente con alta mortalidad que, a menudo, es subdiagnosticada. El diagnóstico temprano es fundamental porque la intervención oportuna mejora el pronóstico del paciente. Existen tres tipos de síndrome hemofagocítico según su causa desencadenante: primario (genético), secundario (infeccioso, reumatológico, oncológico) y de origen desconocido.4,5,6 En el contexto de infecciones, el virus de Epstein-Barr es el agente más comúnmente asociado.6 La manifestación clínica inicial incluye fiebre persistente y hepatoesplenomegalia, que puede evolucionar hacia cuadros críticos similares a sepsis.7,8
El diagnóstico se facilita mediante los criterios clínico-analíticos establecidos en el protocolo HLH-2004, que incluyen: fiebre persistente, esplenomegalia, citopenias, hipertrigliceridemia, hipofibrinogenemia, hiperferritinemia, CD25 soluble elevado, disminución de la actividad citotóxica de las células natural killer y evidencia de hemofagocitosis en la médula ósea, el bazo, los ganglios linfáticos o el hígado. Estos criterios tienen una sensibilidad superior al 99% y especificidad del 80% en la población pediátrica.9
La afectación cutánea se observa en, incluso, el 65% de los casos, con lesiones que van desde exantemas maculopapulares simples hasta eritrodermia, paniculitis y púrpura.6,10 En términos histológicos, se observa un infiltrado linfohistiocítico perivascular en la dermis reticular.11
El tratamiento se basa en las guías HLH-2004 e incluye etopósido, dexametasona y ciclosporina durante una a ocho semanas, con cobertura antimicrobiana de amplio espectro hasta obtener los resultados de los cultivos.12,13 Puede indicarse tratamiento coadyuvante con inmunoglobulina humana cada cuatro semanas.12,13 En casos resistentes los tratamientos inmunomoduladores, como rituximab y ruxolitinib, han mostrado eficacia en algunos pacientes.14,15,16
CONCLUSIONES
El síndrome hemofagocítico representa un desafío diagnóstico y terapéutico porque sus manifestaciones clínicas son diversas y pueden afectar múltiples sistemas (cutáneo, neurológico, pulmonar).17 La identificación temprana de estas manifestaciones, junto con una sospecha clínica elevada, son decisivas para el inicio rápido del tratamiento, lo que puede repercutir significativamente en el pronóstico del paciente.
REFERENCIAS
1. Salunke B, Savarkar S. Hemophagocytic syndrome—An approach to the management. Indian J Crit Care Medicine 2019; 23 (S3). https://doi.org/10.5005/jp-journals-10071-23251
2. Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol 2013; 161: 609-622. https://doi.org/10.1111/bjh.12293
3. Santos IO, Neto RP, Paula A. Hemophagocytic lymphohistiocytosis: a case series analysis in a pediatric hospital. Hematol Transfusion Cell Ther 2021; 45 (1): 32-7. https://doi.org/10.1016/j.htct.2021.04.006
4. Itziar Astigarraga LIG. Haemophagocytic syndromes: the importance of early diagnosis and treatment. Anales Pediatría (English Edition) 2018; 89: 124.e1-124.e8. https//doi.org/10.1016/j.anpede.2018.05.002
5. Sen ES, Steward CG, Ramanan AV. Diagnosing haemophagocytic syndrome. Arch Dis Childhood 2017; 102: 279-284.
6. George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med 2014; 5: 69-86. https://doi.org/10.2147/JBM.S46255
7. Terán Pazmiño EE, Paredes Castillo FP, Martínez Mena JM, et al. Síndrome hemofagocítico en niños: Reporte de caso. Rev Ecuatoriana Pediatr 2020; 21 (2): 1-8. https://doi.org/10.52011/0005
8. Morimoto A, Nakazawa Y, Ishii E. Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management. Pediatr Int 2016; 58: 817-825. https://doi.org/10.1111/ped.13064
9. Debaugnies F, Mahadeb B, Ferster A, et al. Performances of the H-score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients. Am J Clin Pathol 2016; 145 (6): 862-70. https://doi.org/10.1093/ajcp/aqw076
10. Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology 2015; 230 (3): 234-243. https://doi.org/10.1159/000368552
11. Morrell DS, Pepping MA, Scott JP, et al. Cutaneous manifestations of hemophagocytic lymphohistiocytosis. Arch Dermatol 2002; 138 (9): 1208-1212. https://doi.org/10.1001/archderm.138.9.1208
12. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood Cancer 2007; 48 (2): 124-131. https://doi.org.10.1002/pbc.21039
13. Bergsten E. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood 2017; 130: 2728-2738. https://doi.org/10.1182/blood-2017-06-788349
14. Chellapandian D, Wiener S, Zelley K, et al. Use of the anti-CD20 antibody rituximab in the treatment of Epstein-Barr virus-induced hemophagocytic lymphohistiocytosis. Blood 2011; 118 (21): 2178. https://doi.org/10.1182/blood.V118.21.2178.2178
15. Shahzad M, Sarfraz Z, Azam R, et al. Efficacy of ruxolitinib in patients with hemophagocytic lymphohistiocytosis: A systematic review. Blood 2024; 144 (Supplement 1): 5372. https://doi.org/10.1182/blood-2024-201977
16. Keenan C, Nichols KE, Albeituni S. Use of the JAK inhibitor ruxolitinib in the treatment of hemophagocytic lymphohistiocytosis. Front Immunol 2021; 12: 614704. https://doi.org.10.3389/fimmu.2021.614704
17. Jovanovic A, Kuzmanovic M, Kravljanac R, et al. Central nervous system involvement in hemophagocytic lymphohistiocytosis: a single-center experience. Pediatr Neurol 2014; 50: 233-237.
Este artículo debe citarse como: Izabal-Tinajero N, González-López E, Zepeda-Muñoz E, Palacios-Vega P. Cuando la piel habla: manifestaciones cutáneas como primera señal de un síndrome hemofagocítico de origen desconocido. Dermatol Rev Mex 2025; 69 (4): 603-606.
