Chronic urticaria: Vasculitis vs lupus? A great clinical dilemma.
Dermatol Rev Mex. 2021; 65 (4): 665-671. https://doi.org/10.24245/dermatolrevmex.v65i4.6620
Luis Francisco Pineda-Galindo,1,3 Giselle Madaí Zamacona-Damián,1,3 Mitzi Gabriela Márquez-Vargas,1,3 Orestes de Jesús Cobos-Quevedo,1,3 Itzel María Montoya-Fuentes,2 Cynthia Mireya Rentería-Guevara1,3
1 Departamento de Medicina Interna.
2 Departamento de anatomía patológica.
UMAE-Especialidades Dr. Antonio Fraga Mouret, CMN La Raza, Instituto Mexicano del Seguro Social, Ciudad de México.
3 División de Estudios de Posgrado, Facultad de Medicina. Universidad Autónoma de México, Ciudad de México.
ANTECEDENTES
La urticaria es una manifestación dermatológica frecuente, del 15 al 20% de la población la padece una vez en la vida y del 0.1 al 3% tendrán una forma crónica durante más de seis semanas.1 La urticaria recurrente es un trastorno que a menudo representa un desafío para su diagnóstico y tratamiento. La causa del 60% de los casos de urticaria recurrente es idiopática y sólo el 10% de los pacientes tienen vasculitis urticariana, que puede dividirse en dos grupos, normocomplementémica e hipocomplementémica. La mayoría de los casos de vasculitis urticariana normocomplementémica son idiopáticos, a diferencia de la hipocomplementémica que puede ser secundaria a enfermedades, como lupus eritematoso sistémico, síndrome de Sjögren y neoplasias, entre otras. La vasculitis urticariana hipocomplementémica también puede ser primaria, en tal caso se define como síndrome de vasculitis urticariana hipocomplementémica.2
La vasculitis urticariana hipocomplementémica es una afección anatomoclínica definida por el daño inflamatorio de los capilares de la dermis asociado con una erupción urticariana atípica.3 Se calcula una incidencia global de 0.7 por millón de habitantes, comparable a la de otras vasculitis de vasos pequeños y medianos, como la poliarteritis nodosa y la granulomatosis eosinofílica con poliangeítis.4
Es una afección rara, descrita por McDuffie en 1973 en un informe de cuatro pacientes con lesiones cutáneas asociadas con concentraciones bajas de complemento, en los que se descartaron otras enfermedades del tejido conectivo (crioglobulinemia, lupus eritematoso sistémico, etc). Comprende una reacción de hipersensibilidad de tipo III mediada por depósitos de inmunocomplejos en los capilares y las vénulas poscapilares.5,6
En términos clínicos, se manifiesta con habones que, de forma característica, pueden contener focos purpúricos a lo largo de su evolución. Con frecuencia se acompañan de angioedema y síntomas sistémicos con afectación articular, ocular, pulmonar o digestiva. El estudio histopatológico es compatible con vasculitis leucocitoclástica de los vasos de pequeño calibre dérmicos e importante edema en la dermis.7
El tratamiento se enfoca en reducir la inflamación y lesiones en los órganos afectados.8
Se comunica una revisión de la bibliografía a propósito de dos casos de pacientes con urticaria crónica y manifestaciones sistémicas en los que se sospechó lupus eritematoso sistémico y en quienes el diagnóstico definitivo fue vasculitis urticariana hipocomplementémica.
CASOS CLÍNICOS
Caso 1
Paciente femenina de 42 años, antecedente de migraña en tratamiento sintomático. Inició su padecimiento en diciembre de 2019 con urticaria crónica resistente, valorada por el servicio de Dermatología que otorgó tratamiento con epinastina sin respuesta, posteriormente se consideró lupus eritematoso sistémico con actividad cutánea por lo que se agregó cloroquina; la paciente tuvo mejoría parcial; sin embargo, cursó con dermatosis caracterizada por ronchas palpables, eritematosas, con hiperemia, en la región preauricular izquierda, con posterior generalización de placas confluentes en la cara, el cuello y el tronco, así como en las extremidades agravadas por exposición solar, además de prurito y artralgias en el codo y la rodilla derechas. Figura 1
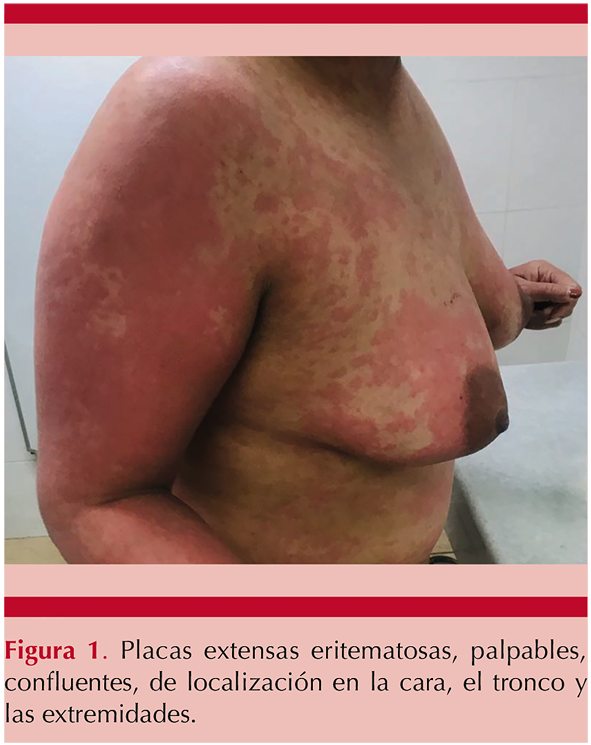
Ingresó a cargo del servicio de Medicina Interna para protocolo de estudio con sospecha de actividad cutánea por lupus eritematoso sistémico. Los estudios iniciales no mostraron afección hematológica o bioquímica, con serología viral (TORCH, VHC, VHB, VIH) negativa; en el perfil reumatológico destacó hipocomplementemia con reporte de C4 en 8 mg/dL (16-38 mg/dL), C3 de 57 mg/dL (79-152 mg/dL) y anticuerpos antinucleares positivos 1:80 con patrón moteado fino, pero el resto de los autoanticuerpos, anti-Sm, anti-La, anti- Ro y anti-ADN doble cadena negativos.
Se realizó biopsia en sacabocado de las lesiones del hombro derecho y la pierna izquierda, con reporte de infiltrado inflamatorio crónico linfocitario leve no específico perivascular y perianexial con edema focal en la dermis superficial. Figura 2
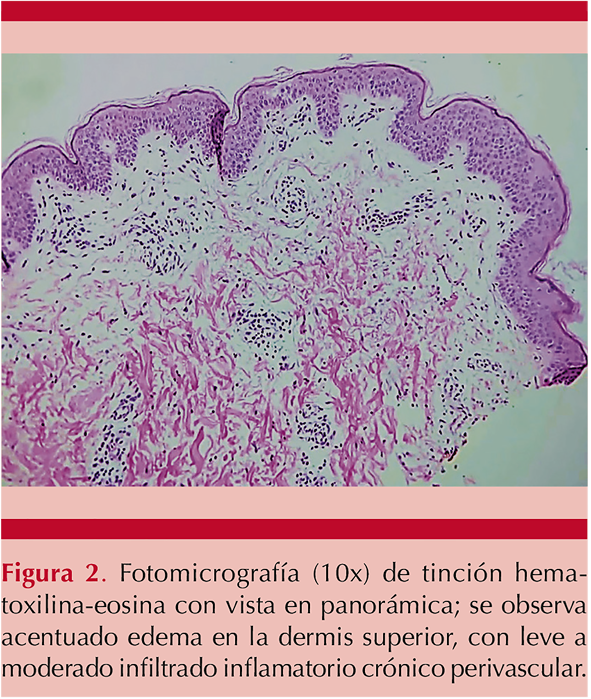
Ante estos hallazgos, con evidencia de disminución de las concentraciones de complemento, la presencia de ANA positivo, con negatividad de anti-ADN y ausencia de datos clínicos compatibles con lupus eritematoso sistémico, se estableció el diagnóstico definitivo de vasculitis urticariana hipocomplementémica. Se inició tratamiento con esteroide intravenoso a dosis de 0.5 mg/kg de peso al día (hidrocortisona al día durante 3 días) aunado a hidroxizina, seguido de prednisona con dosis de reducción. La paciente tuvo desaparición de las lesiones urticarianas que dejaron máculas hiperpigmentadas que tras varias semanas desaparecieron, así como las artralgias; finalmente la paciente egresó por mejoría con seguimiento ambulatorio.
Caso 2
Paciente femenina de 25 años que tenía el antecedente de lesiones cutáneas compatibles con urticaria desde la adolescencia, con tratamiento con hidroxizina sin respuesta; con diarrea acuosa en los últimos tres años, de manera episódica, sin relación con alimentos y de alivio espontáneo. Desde hacía seis meses la paciente manifestó artralgias generalizadas y aparente rigidez matutina. Tuvo exacerbación del cuadro diarreico con intolerancia a la vía oral, lo que condicionó su hospitalización.
Al examen físico mostró dermatosis diseminada a la cara, el tronco y las extremidades, caracterizada por habones confluentes que formaban placas eritematosas, pruriginosas, con persistencia por más de 72 horas, que evolucionaban a máculas eritematosas y purpúricas, algunas con hiperpigmentación posinflamatoria. Figura 3
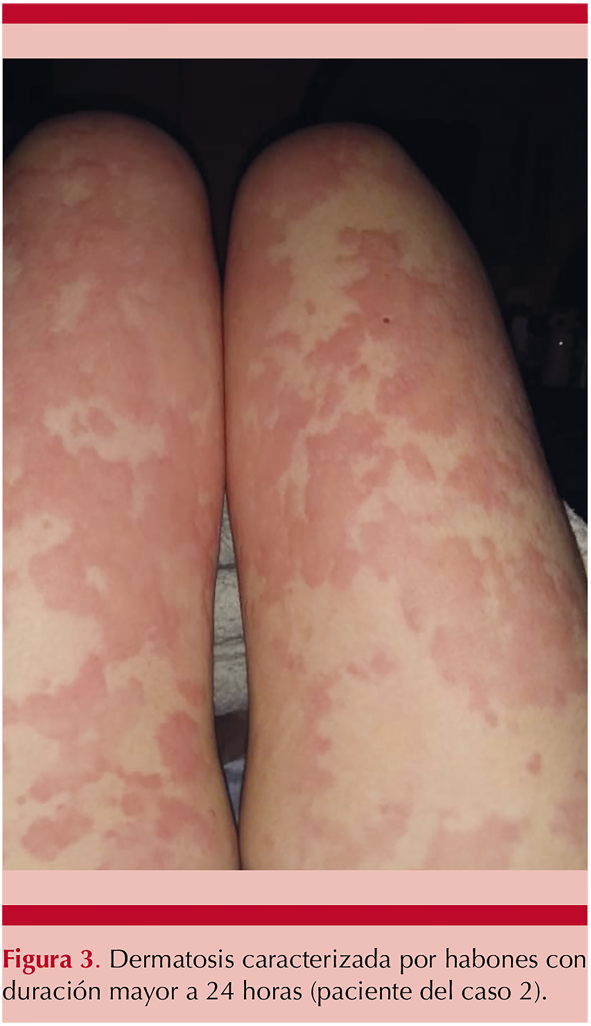
Se encontró anemia normocítica normocrómica (Hb 9.9 g/dL), elevación de creatinina (3.53 mg/dL), proteinuria subnefrótica (1.07 g en 24 horas), positividad de anticuerpos antinucleares a dilución 1:160 patrón moteado fino y actividad del complemento disminuida, con C4 en 4 mg/dL (16-38 mg/dL) y C3 de 48 mg/dL (79-152 mg/dL), y C1q inhibidor de 34 mg/dL (21-39 mg/dL) con serología viral (TORCH, VIH, VHC, VHB) negativa y anticuerpos anti-ADN de doble cadena, ANCAS, anti-Sm, anti-La y anti-Ro negativos. Se realizó biopsia de las lesiones cutáneas y el reporte histopatológico reveló vasculitis leucocitoclástica. Figura 4
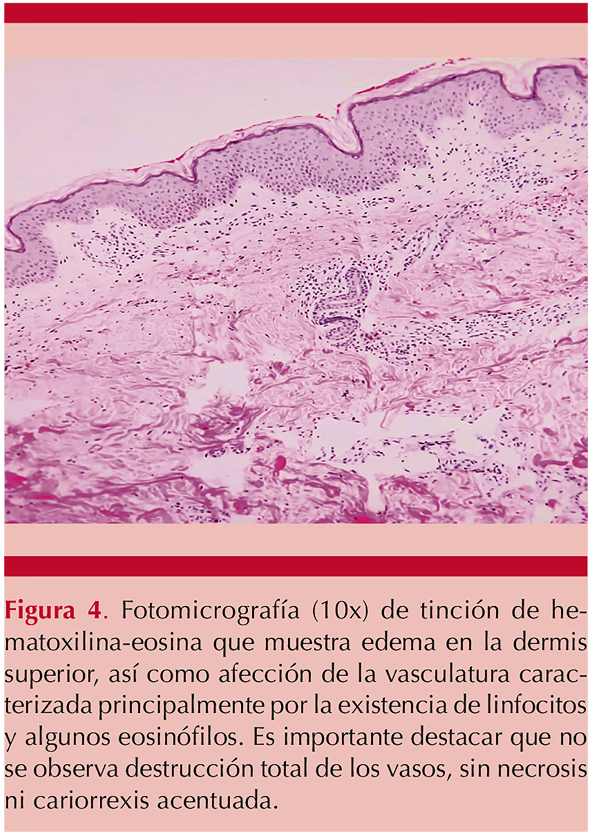
En este contexto, se sospechó de manera inicial ante las alteraciones serológicas de complemento sérico bajo, ANA positivo y daño renal manifestado por proteinuria la posibilidad de lupus eritematoso sistémico, por lo que se trató con esteroide a dosis de 1 mg/kg al día, seguido de una dosis de reducción de prednisona (0.5 mg/kg al día), ante el resultado negativo de los anti-ADN, anti- Sm y otros autoanticuerpos se descartó ese diagnóstico y se definió como una vasculitis urticariana hipocomplementémica tras el tratamiento con remisión de los síntomas, las lesiones dérmicas y la afección renal y gastrointestinal.
DISCUSIÓN
La vasculitis urticariana hipocomplementémica es una enfermedad infrecuente, poco vista en la práctica médica, incluso en un servicio de medicina interna de tercer nivel, sus manifestaciones clínicas pueden confundirse con otras afecciones, lo que la hace un padecimiento infradiagnosticado. Tiene prevalencia aproximada del 5% y es más frecuente en mujeres (60 al 80% de los casos), su incidencia alcanza su punto máximo en la cuarta década de la vida. La edad promedio de manifestación es de 48 años.9
La principal manifestación es una erupción cutánea o artralgias durante meses previos al diagnóstico, como fue el caso de ambas pacientes, quienes permanecieron con estos síntomas durante nueve meses en el caso 1 y hasta más de 10 años antes del diagnóstico definitivo en el caso 2. Las lesiones cutáneas pueden ser dolorosas, acompañadas de sensación de quemadura y prurito. La característica más importante es la persistencia de la lesión cutánea durante más de 24 horas, lo que sugiere que se trata de una vasculitis leucocitoclástica o linfomonocítica y no de una urticaria no vasculítica, que es de pocas horas de duración y nunca permanece más de 24 horas.10 Las manifestaciones extracutáneas incluyen síntomas constitucionales (fiebre, malestar y fatiga), síntomas músculo-esqueléticos (artralgias y artritis, las más frecuentes), conjuntivitis, epiescleritis y uveítis, serositis, enfermedad pulmonar obstructiva, enfermedad renal, síntomas gastrointestinales (dolor abdominal, náusea, vómito, diarrea, ascitis, hepatomegalia, esplenomegalia), daño cardiaco (pericarditis, valvulopatías), entre otros.2,11
El diagnóstico se basa en los criterios de Schwarz (Cuadro 1); se define con la existencia de ambos criterios mayores y dos de los seis criterios menores.12

Nuestras pacientes cumplieron con ambos criterios mayores y dos de los criterios menores (vasculitis leucocitoclástica en biopsia de piel y artralgias) con lo que establecimos el diagnóstico definitivo. En el segundo caso no pudo definirse si la afección renal fue secundaria a la vasculitis o a la deshidratación.
La biopsia de lesiones cutáneas activas es el patrón de referencia para el diagnóstico de esta enfermedad. En ella se evidencia vasculitis de vasos pequeños, que se asocia con leucocitoclasia (fragmentación de leucocitos con detritus nucleares o “polvillo nuclear”), edema endotelial y destrucción de la pared vascular con depósito de fibrinoide, eventual extravasación de glóbulos rojos e infiltración granulocítica perivascular; este hallazgo no es patognomónico porque encontramos este patrón histopatológico secundario a fármacos, infecciones, conectivopatías, neoplasias o en forma idiopática.13
Los hallazgos bioquímicos más comunes en la vasculitis urticariana son aumento de la velocidad de sedimentación globular, hipocomplementemia (generalmente C3 y C4), inmunocomplejos circulantes y ANA positivo. La hipocomplementemia es un marcador aceptado de enfermedad sistémica y se asocia con más complicaciones en el curso de la enfermedad.7
Como parte del diagnóstico diferencial, la consideración de encontrarse ante lupus eritematoso sistémico continúa siendo un reto clínico porque está reportado que la vasculitis urticariana hipocomplementémica está presente en el 7 al 8% de los pacientes con lupus eritematoso sistémico y el 54% de los pacientes con vasculitis urticariana hipocomplementémica son diagnosticados con lupus eritematoso sistémico en el periodo de seguimiento. Sin embargo, la erupción típica en mariposa no ocurre en pacientes con vasculitis urticariana hipocomplementémica, y las ronchas y angioedema son raros en el lupus eritematoso sistémico. Asimismo, aproximadamente el 50% de los pacientes con vasculitis son positivos para anticuerpos antinucleares, aunque los anticuerpos anti-ADN rara vez son positivos, éste es el principal criterio bioquímico diferencial con lupus eritematoso sistémico; en ambos casos la negatividad de los anticuerpos anti-ADN de doble cadena, los síntomas y el reporte de la biopsia hizo descartar el diagnóstico de lupus eritematoso sistémico. Los anticuerpos anti-C1q son positivos en el 30% de los pacientes con lupus eritematoso sistémico, así como en otras enfermedades autoinmunitarias, como el síndrome de Sjögren.14,15
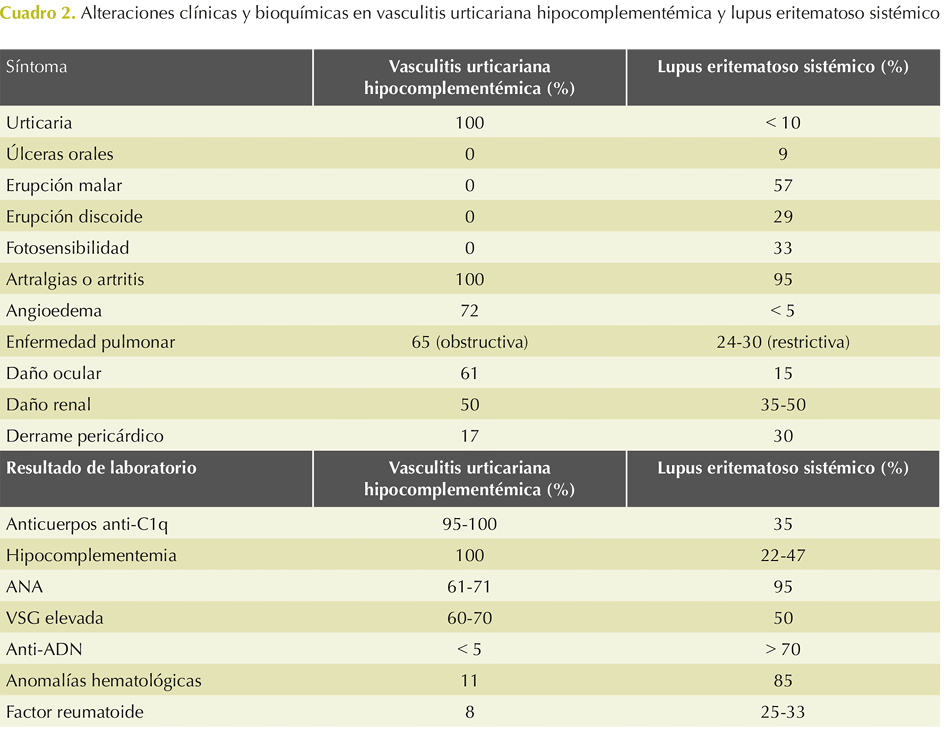
El Cuadro 2 muestra la frecuencia de las manifestaciones clínicas y bioquímicas entre el principal diagnóstico diferencial de la vasculitis urticariana hipocomplementémica.
Debido a la baja frecuencia de la enfermedad y su infradiagnóstico, no se cuenta con guías establecidas para su tratamiento. Los corticosteroides son los más prescritos y conducen a la remisión de las lesiones cutáneas en más del 80%. En casos limitados a la piel se han administrado hidroxicloroquina, dapsona, colchicina con respuesta satisfactoria, mientras que en aquéllos con enfermedad sistémica se han prescrito agentes como metotrexato, azatioprina, micofenolato, ciclofosfamida, tocilizumab, factor anti-IL1, ciclosporina y rituximab. En pacientes con enfermedad más grave, la combinación de esteroides y ciclofosfamida es el tratamiento de primera línea.7 El pronóstico de la vasculitis urticariana hipocomplementémica depende del tipo de órgano afectado, la gravedad se relaciona principalmente con el daño pulmonar y renal. Por último, incluso si la vasculitis urticariana hipocomplementémica representa una enfermedad sistémica, crónica y recurrente, el pronóstico es satisfactorio, con buena supervivencia global. Las muertes directamente atribuibles a la enfermedad son excepcionales.
Por otro lado, las complicaciones graves de origen infeccioso o cardiovascular son frecuentes y parecen estar relacionadas en su mayor parte con el tratamiento con esteroides de larga duración y otros tratamientos sistémicos.14
En conclusión, la vasculitis urticariana hipocomplementémica es una vasculitis de pequeño vaso poco frecuente, con múltiples manifestaciones clínicas, que fácilmente puede confundirse con diferentes afecciones, particularmente con lupus eritematoso sistémico, diagnóstico diferencial obligado. En ello radica la importancia de su conocimiento, no sólo entre especialistas en diferentes ramas (medicina interna, reumatología, dermatología, entre otras), sino también entre médicos de primer contacto, limitando de esta manera tratamientos ineficaces; su diagnóstico oportuno permite el tratamiento dirigido que influye en la calidad de vida del paciente, la repercusión a órganos y sistemas y, por tanto, en el pronóstico.
REFERENCIAS
- Vigan M. Urticarias. EMC–Tratado de medicina. 2017; 21 (4): 1-7. doi. 10.1016/S1636-5410(17)86936-1.
- Jara LJ, Navarro C, Medina G, Vera-Lastra O, et al. Hypocomplementemic urticarial vasculitis syndrome. Curr Rheumatol Rep 2009; 11 (6): 410-5. doi. 10.1007/s11926-009-0060-y.
- Jachiet M, Flageul B, Bouaziz JD, Bagot M, et al. Les vascularites urticariennes hypocomplémentémiques. Rev Med Interne 2018; 39 (2): 90-8. doi. 10.1016/j.revmed.2017.03.005.
- Sjöwall C, Mandl T, Skattum L, Olsson M, et al. Epidemiology of hypocomplementaemic urticarial vasculitis (anti-C1q vasculitis). Rheumatology (Oxford) 2018; 57 (8): 1400-7. doi. 10.1093/rheumatology/key110.
- Alomari M, Al Momani L, Khazaaleh S, Almomani S, et al. Exceptional association of hypocomplementemic urticarial vasculitis syndrome (HUVS) and symptomatic pulmonary histoplasmosis: a case-based literature review. Clin Rheumatol 2019; 38 (6): 1691-7. doi. 10.1007/s10067-019-04548-8.
- Vallianou K, Skalioti C, Liapis G, Boletis JN, et al. A case report of hypocomplementemic urticarial vasculitis presenting with membranoproliferative glomerulonephritis. BMC Nephrol 2020; 21 (1): 351. doi. 10.1186/s12882-020-02001-6.
- Pulido-Pérez A, Avilés-Izquierdo JA, Suárez-Fernández R. Vasculitis cutáneas. Actas Dermosifiliogr 2012; 103 (3): 179-91. doi. 10.1016/j.ad.2011.06.001.
- González SA, Muñoz C, Moncada AP. Urticaria vasculítica: Un diagnóstico diferencial amplio y poco frecuente. Piel (Barc) 2019; 34 (4): 2-6. doi. 10.1016/j.piel.2018.10.001.
- Moreno-Suárez F, Pulpillo-Ruiz Á, Zulueta-Dorado T, Conejo-Mir Sánchez J. Urticaria vasculitis: estudio retrospectivo de 15 casos. Actas Dermosifiliogr 2013; 104 (7): 579-85. doi. 10.1016/j.ad.2012.12.004.
- Maldonado JE, Iglesias-Gamarra A. Vasculitis urticarial hipocomplementémica: aclaración histórica. Rev Colomb Reumatol 2014; 21 (2): 84-90. doi. 10.1016/S0121-8123(14)70153-7.
- Hauser B. Systemic manifestations of hypocomplementemic urticarial vasculitis: Comment on the article by Jachiet, et al. Arthritis Rheumatol 2015; 67 (7): 1984-5. doi. 10.1002/art.39097.
- Jachiet M, Flageul B, Deroux A, Le Quellec A, et al. The clinical spectrum and therapeutic management of hypocomplementemic urticarial vasculitis: data from a French nationwide study of fifty-seven patients. Arthritis Rheumatol 2015; 67 (2): 527-34. https://doi.org/10.1002/art.38956.
- Ion O, Obrișcă B, Ismail G, Sorohan B, et al. Kidney involvement in hypocomplementemic urticarial vasculitis syndrome-A case-based review. J Clin Med 2020; 9 (7): 2131. doi. 10.3390/jcm9072131.
- Parra V, Aguirre HD, Daza RA, Mora SA, et al. Lupus eritematoso sistémico versus urticaria vasculítica hipocomplementémica: un dilema diagnóstico en la práctica clínica. Rev Colomb Reumatol 2015; 22 (3): 168-73. doi. 10.1016/j.rcreu.2015.05.003.
- Smith EMD, Lythgoe H, Hedrich CM. Vasculitis in juvenile-onset systemic lupus erythematosus. Front Pediatr 2019; 7 (149): 1-9. doi. 10.3389/fped.2019.00149.
Recibido: mayo 2021
Aceptado: mayo 2021
Este artículo debe citarse como: Pineda-Galindo LF, Zamacona-Damián GM, Márquez-Vargas MG, Cobos-Quevedo OJ, Montoya-Fuentes IM, Rentería-Guevara CM. Urticaria crónica: ¿vasculitis vs lupus? El gran dilema clínico. Dermatol Rev Mex 2021; 65 (4): 665-671.
