The case of Edward Lambert “the porcupine man”: History and update of a singular dermatosis.
Dermatol Rev Mex. 2025; 69 (2): 277-281. https://doi.org/10.24245/dermatolrevmex.v69i2.10450
Daira Ixchel Velazco Muciño,1 Marcela Hernández Vera,2 María Elisa Vega Memije2
1 Estudiante de Medicina, Facultad de Medicina, UNAM.
2 División de Dermatología, Hospital General Dr. Manuel Gea González, Ciudad de México.

Descripción de una nueva enfermedad cutánea según John Machin
En 1731, a la edad de 14 años, Edward Lambert, quien padecía una peculiar afección dermatológica, fue presentado por su padre a John Machin, secretario de la Royal Society de Londres, quien, asombrado por el caso, publicó en 1732 un informe en Philosophical Transactions, en el que detalló las manifestaciones clínicas de la enfermedad.1
En este informe describió la piel de Lambert como “una funda gruesa de color oscuro, que se ajustaba perfectamente a su cuerpo, hecha de una corteza o piel rugosa, con cerdas en algunos lugares, que cubría toda la superficie corporal, excepto la cara, las palmas y las plantas, dando la impresión de que sólo esas partes estaban desnudas y el resto cubiertas”.1
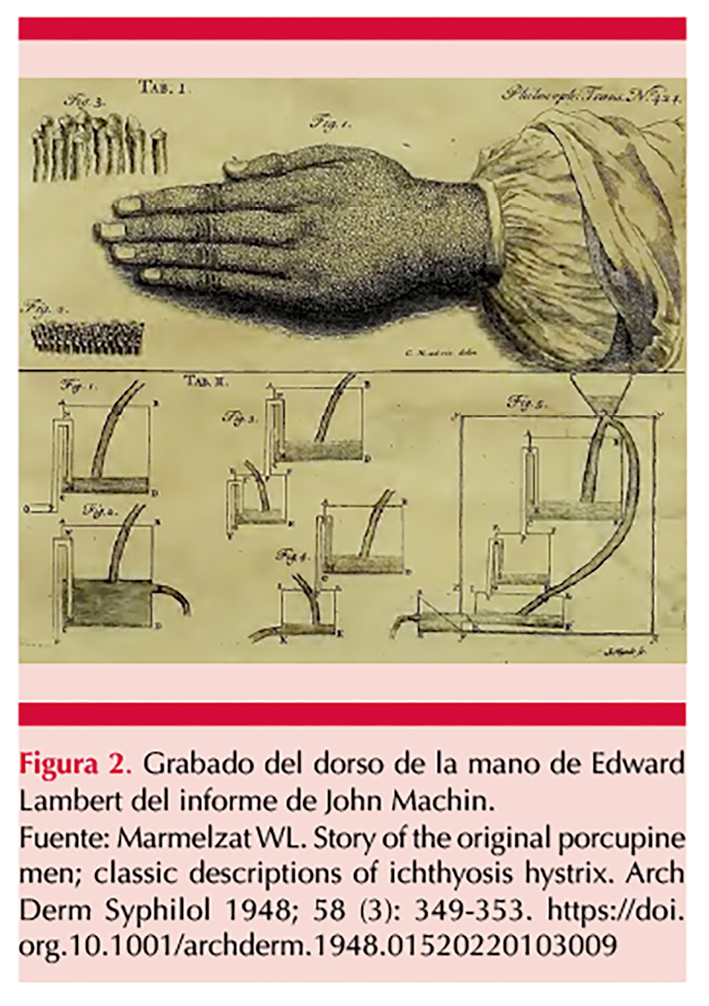
John Lambert, descendiente de Edward, fue retratado en un grabado por Wilhelm, quien ilustró la distribución topográfica de la dermatosis (Figura 1).2 Además, en su reporte, Machin incluyó un grabado que representa la superficie cutánea del dorso de la mano de Edward (Figura 2). En su descripción, detalló cómo la ilustración mostraba, a gran aumento, las “papilas nerviosas” que sobresalían de la epidermis, distribuidas de manera uniforme, de tamaño homogéneo y adoptando diversas formas.1
Se negó que algún accidente hubiera causado esta afección, así como la existencia de enfermedades cutáneas similares en otros integrantes de la familia. Mencionó que la alteración cutánea surgió cuando Edward tenía siete u ocho semanas de vida, en el momento en que su piel comenzó a tornarse amarilla, posteriormente negra y a engrosar con el tiempo. Cada año, en otoño, al alcanzar un grosor de 1.9 cm, ésta se desprendía y era sustituida por una nueva con las mismas características.1
Agregó que, a pesar de la enfermedad, su desarrollo físico y bienestar no se veían afectados: “el tamaño era apropiado para su edad; su cuerpo y extremidades eran rectos y bien formados”, “esta cubierta áspera no le causaba dolor ni malestar, excepto que a veces, después de un trabajo duro, tendía a romperse y a sangrar”.1
Publicaciones complementarias del caso Lambert
El caso atrajo la atención de otros autores contemporáneos; uno de ellos fue Baker, quien en 1755 detalló cómo la piel de Edward Lambert simulaba “verrugas cilíndricas, rígidas y elásticas, que crujían al pasar la mano sobre ellas”, por lo que le acuñó el seudónimo de “el hombre puercoespín”. Además, señaló que Edward tuvo seis hijos, incluidos hombres y mujeres; sin embargo, la afección sólo se manifestó en los varones. También especuló sobre el origen de una nueva “raza” si este trastorno se perpetuaba generacionalmente.1
Otro autor fue Ascanius, quien señaló que, durante muchos años, Edward Lambert y sus descendientes se ganaron la vida exponiéndose públicamente en ferias de Europa.2,3 Figura 3

En 1806, a la edad de 90 años, Lambert falleció a causa de un accidente, lo que demuestra que su esperanza de vida no se redujo por su enfermedad.3 Este trastorno cutáneo se reportó en 11 miembros de la familia en cuatro generaciones consecutivas, hasta 1834, cuando el último miembro afectado de la familia murió en la infancia.1,2
DISCUSIÓN
La ictiosis hystrix (IH) engloba a un grupo heterogéneo de trastornos de la queratinización poco común, que se distinguen por crestas hiperqueratósicas y verrugosas de coloración marrón oscuro. Su nombre proviene de las palabras griegas “ichthyos” (pez) e “hystrix” (puercoespín).4-7 El término de ictiosis hystrix también se utiliza para referirse a un tipo de nevo epidérmico con distribución bilateral.5
Se han descrito cinco variantes clínicas: Brocq, Lambert, Curth-Macklin, Rheydt y Bäfverstedt. El patrón de distribución topográfica de las lesiones, así como las anomalías asociadas y los patrones de herencia, ayudan a diferenciar entre estos subtipos.4,6,8
La ictiosis tipo Lambert, enfermedad que padeció Edward Lambert, se caracteriza por diseminación a todos los segmentos corporales, excepto la cara, los genitales, las palmas y las plantas. El tipo Curth-Macklin se distingue por queratodermia palmoplantar con escamas gruesas, a menudo espinosas. La principal característica diferencial respecto a la ictiosis tipo Lambert es que la ictiosis tipo Curth-Macklin afecta la piel de las palmas, las plantas o ambas. En el tipo Brocq, es común la eritrodermia y las ampollas, mientras que las formas nevoides son menos frecuentes. El tipo Rheydt causa hiperqueratosis en la cara y las extremidades, junto con pérdida de la audición. Finalmente, el tipo Bäfversted afecta especialmente la cara, con una hiperqueratosis folicular prominente, mientras que las palmas se ven levemente afectadas.4,6-10
Revisión de 1958 sobre la ictiosis tipo Lambert: patrón de trasmisión y variabilidad clínica
En 1958, Penrose y Stern publicaron una actualización de la ictiosis tipo Lambert; demostraron que esta enfermedad también afectó a las mujeres. Inicialmente se sugirió una asociación con el gen Y; sin embargo, los autores refutaron esta hipótesis y concluyeron que el patrón de herencia es autosómico dominante. Del mismo modo, describieron la existencia de variaciones en la gravedad de las lesiones entre los integrantes de una misma familia según el sexo, la edad y la localización anatómica, así como la estación del año. Señalaron que, posiblemente, casos muy leves pudieron haber pasado inadvertidos.2
Casos comunicados a lo largo del tiempo y nuevas actualizaciones de la ictiosis tipo Lambert
Tras la descripción del “hombre puercoespín”, se han descrito pocos casos compatibles con ictiosis tipo Lambert: los reportados por Capetanakis y su grupo en 1975 y por Kriner y Montes de Argentina en 1997.4,6 En 1993, Niemi y colaboradores y Kanerva y su grupo describieron una familia de cuatro integrantes, incluidas mujeres, en tres generaciones, a quienes se les diagnosticó ictiosis tipo Curth-Macklin. No obstante, Wang y su grupo señalaron que las características cutáneas y el comportamiento clínico de la enfermedad eran concordantes con ictiosis tipo Lambert.4
En 2007, Wang y su grupo comunicaron el caso de ictiosis tipo Lambert en tres pacientes de dos generaciones de una familia china. En 2016 se publicó una actualización en la que se propuso unificar la ictiosis tipo Lambert y la tipo Curth-Macklin como una sola afección, diferenciada por las mutaciones en los genes KRT1 y KRT10 (queratina 1 y 10). Las mutaciones en KRT1 se asociaron con afectación palmoplantar, característica de la ictiosis tipo Curth-Macklin, mientras que en la alteración de KRT10 no hubo afectación de estas áreas, hallazgos típicos de ictiosis tipo Lambert.4,7,10
Sin embargo, en 2018, Terrinoni y su grupo refutaron la hipótesis de Wang y colaboradores al demostrar que la ictiosis tipo Curth-Macklin se asocia con mutaciones KRT1 y KRT10. Por lo tanto, aún no existe una clasificación clara y aceptada de este grupo de ictiosis.7
CONCLUSIONES
La afección dermatológica de Edward Lambert, conocida como ictiosis hystrix tipo Lambert, despertó el interés de la comunidad científica y de la sociedad de la época debido a sus características inéditas. Este caso no sólo impulsó investigaciones de autores contemporáneos, sino que, hasta la fecha, continúa siendo objeto de estudio, enfocado en comprender su origen, los factores genéticos implicados y el patrón de trasmisión hereditaria. Después de la familia Lambert, sólo se ha comunicado una cantidad limitada de casos adicionales, en los que los resultados no son concluyentes.
REFERENCIAS
1. Marmelzat, W L. Story of the original porcupine men; classic descriptions of ichthyosis hystrix. Arch Derm Syphilol 1948; 58 (3): 349-353. https://doi.org.10.1001/archderm.1948.01520220103009
2. Penrose LS, Stern C. Reconsideration of the Lambert pedigree (ichthyosis hystrix gravior). Ann Hum Genet 1958; 22 (3): 258-283. https://doi.org.10.1111/j.1469-1809.1958.tb01421.x
3. Traupe, H. (1989). History of the Ichthyoses. In: The Ichthyoses. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-73650-6_2
4. Wang WH, Li LF, Zhang Q, et al. Ultrastructural features of ichthyosis hystrix strongly resembling Lambert type. Br J Dermatol 2007; 156 (5): 1027-1031. https://doi.org.10.1111/j.1365-2133.2007.07792.x
5. Nayak S, Acharjya B, Mohanty P. Ichthyosis hystrix. Indian Dermatol Online J 2013; 4 (1): 47-49. https://doi.org.10.4103/2229-5178.105483
6. Kerry Knight L, McGrath J, Ozoemena L, et al. “A new heterozygous frameshift variant in keratin 10 resulting in ichthyosis hystrix in a father and daughter.” JAAD Case Rep 2023; 35: 22-24. https://doi.org.10.1016/j.jdcr.2023.02.015
7. Terrinoni A, Didona B, Caporali S, et al. Role of the keratin 1 and keratin 10 tails in the pathogenesis of ichthyosis hystrix of Curth Macklin. PLoS One 2018; 13 (4): e0195792. https://doi.org.10.1371/journal.pone.0195792
8. Mehta S, Agarwal US, Agarwal N. A sporadic case of ichthyosis Curth Macklin: Rare presentation of a rare disease. Indian J Dermatol 2015; 60 (5): 522. https://doi.org.10.4103/0019-5154.164439
9. Nair PA, Singhal R, Gandhi S, Diwan N. A sporadic case of ichthyosis hystrix: Curth and Macklin type. Indian Dermatol Online J 2017; 8 (2): 139-141. https://doi.org.10.4103/2229-5178.202264
10. Wang WH, Zhang L, Li LF, Sun TT. Ichthyosis hystrix Lambert type and Curth-Macklin type are a single entity with affected (KRT1 mutation) or unaffected (KRT10 mutation) palms and soles?. Eur J Dermatol 2016; 26 (5): 493-495. https://doi.org.10.1684/ejd.2016.2808
https://orcid.org/0009-0006-4539-9731
Recibido: enero 2025
Aceptado: febrero 2025
Este artículo debe citarse como: Velazco-Muciño DI, Hernández-Vera M, Vega-Memije ME. El caso de Edward Lambert “el hombre puercoespín”: historia y actualización de una dermatosis singular. Dermatol Rev Mex 2025; 69 (2): 277-281.
