Schamberg disease: Unusual case.
Dermatol Rev Mex. 2025; 69 (2): 309-312. https://doi.org/10.24245/dermatolrevmex.v69i2.10456
Nathalya Bermúdez,1 Mildred Dorta,2 Sandra Vivas3
1 Residente de tercer año del posgrado de Dermatología.
2 Residente de segundo año del posgrado de Dermatología.
3 Médico internista-dermatólogo. Jefa del Servicio de Dermatología. Profesora titular de la Escuela de Medicina y coordinadora del posgrado de Dermatología.
Ciudad Hospitalaria Dr. Enrique Tejera. Universidad de Carabobo, Valencia, Venezuela.
Las dermatosis purpúricas pigmentarias son un grupo heterogéneo de dermatosis de causa benigna y de alivio espontáneo. De ellas, la enfermedad de Schamberg es la forma más común de manifestación, también se conoce como púrpura progresiva de Schamberg y fue descrita por Jay Frank Schamberg en 1901.1
En términos clínicos se manifiesta con lesiones tipo máculas eritematosas puntiformes que semejan los granos de pimienta de cayena, principalmente en los miembros inferiores y, por lo general, son asintomáticas o levemente pruriginosas. Es de gran importancia su reconocimiento por el dermatólogo para establecer el diagnóstico temprano y oportuno porque existen estudios que establecen su evolución a lesiones neoplásicas linfoproliferativas tipo micosis fungoide.
Se comunica el caso de una paciente de 23 años, natural y procedente de Carabobo. Tenía el antecedente personal de cefalea vascular con administración indiscriminada de antiinflamatorio no esteroide (AINES). Antecedentes familiares no contributorios, fototipo cutáneo III/VI según Fitzpatrick. La paciente acudió a consulta al Servicio de Dermatología de la Ciudad Hospitalaria Dr. Enrique Tejera, Valencia, Carabobo, en mayo de 2022 por padecer máculas eritematosas no pruriginosas de semanas de evolución.
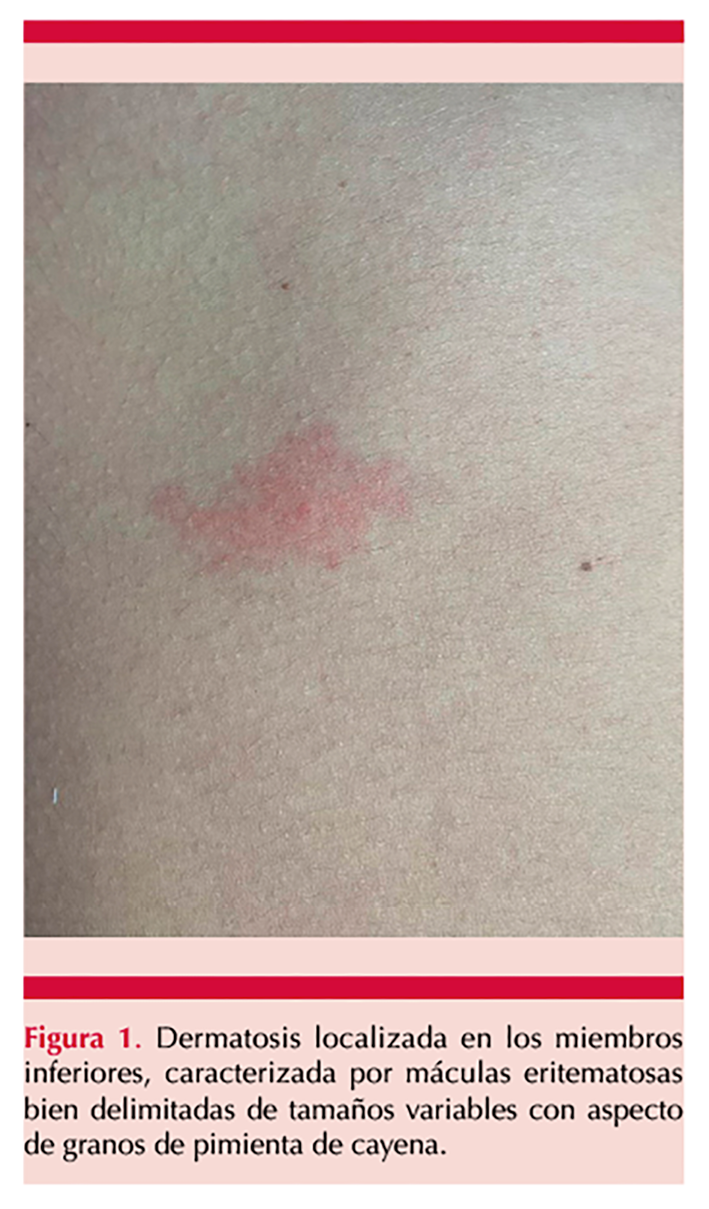
Al examen físico se observó una dermatosis localizada en los miembros inferiores, caracterizada por máculas eritematosas bien delimitadas de tamaños variables con aspecto de granos de pimienta de cayena, no pruriginosa, de tres semanas de evolución, sin afectación palmo-plantar. Figura 1
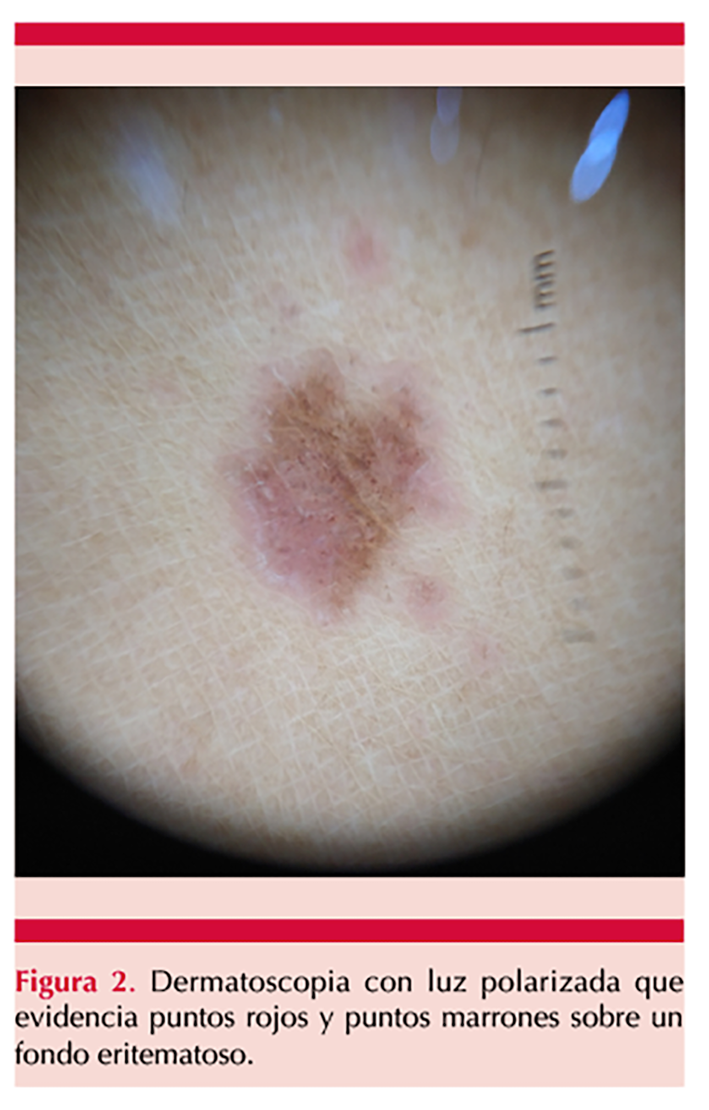
A la dermatoscopia con luz polarizada se evidenciaron puntos rojos y marrones sobre un fondo eritematoso. Figura 2
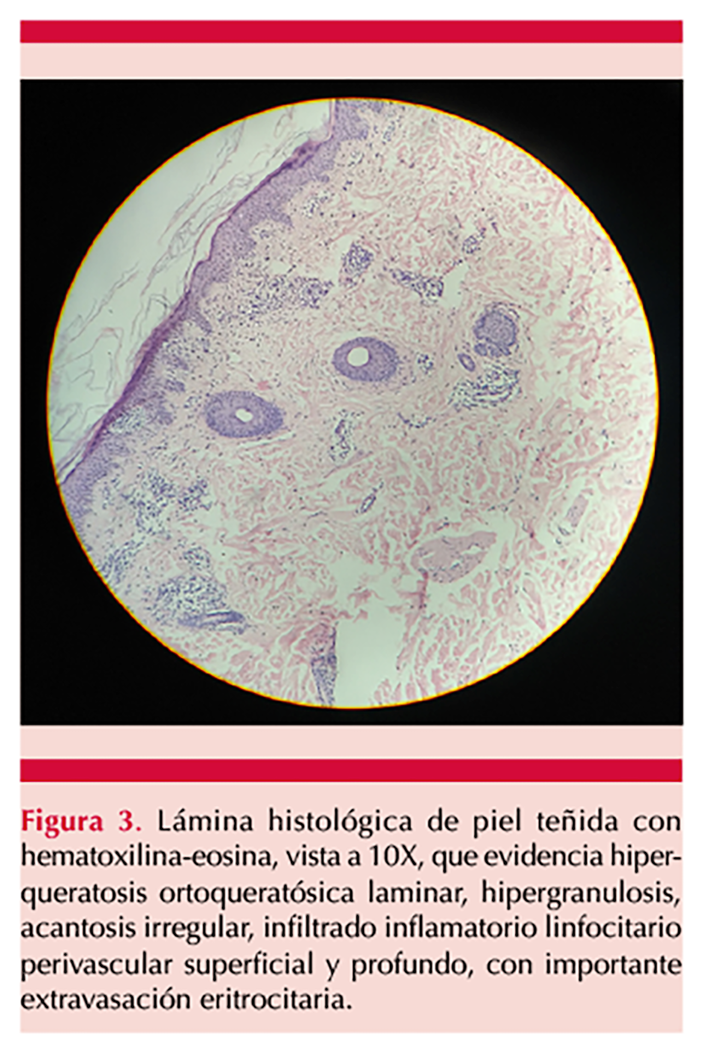
La biometría hemática completa, la química sanguínea, los tiempos de coagulación y serología se encontraban dentro de los límites normales. El estudio de la biopsia cutánea evidenció hiperqueratosis ortoqueratósica laminar, hipergranulosis, acantosis irregular, infiltrado inflamatorio linfocitario perivascular superficial y profundo, con extravasación eritrocitaria. Figura 3
En vista de los hallazgos clínicos, dermatoscópicos e histopatológicos se planteó el diagnóstico definitivo de dermatosis purpúrica pigmentaria progresiva de Schamberg. Se inició tratamiento con medidas generales de la piel, esteroides tópicos de alta potencia (clobetasol al 0.05%), fototerapia UVB- BE y pentoxifilina a dosis de 400 mg al día durante un mes.
Las dermatosis purpúricas pigmentadas son padecimientos poco frecuentes, benignos y de curso crónico, caracterizadas por múltiples petequias sobre máculas hiperpigmentadas pardoamarillentas, que comparten características histopatológicas comunes.1
Clásicamente se han descrito cinco variantes: enfermedad de Schamberg o dermatosis purpúrica pigmentaria progresiva de Schamberg, púrpura pruriginosa o púrpura eccematoide de Doucas y Kapetanakis, dermatosis purpúrica pigmentaria liquenoide de Gougerot y Blum, liquen aureus o purpúrico y púrpura anular telangiectoide o enfermedad de Majocchi.1,2
La dermatosis purpúrica pigmentaria progresiva, o enfermedad de Schamberg, la describió en 1901 Jay Frank Schamberg. Es una enfermedad crónica y recurrente que afecta primordialmente al sexo masculino. No tiene predilección de edad, aunque es muy común en la infancia, en la que se asocia con infecciones virales.2
Su origen es variado y desconocido; tiene múltiples factores desencadenantes: ejercicio físico intenso, diabetes, hipertensión venosa, infecciones, embarazo, fragilidad capilar, fármacos como AINES, ácido salicílico, tiamina, glipizida, interferón, hidralazina, reserpina, pseudoefedrina, bezafibrato y alcohol, entre otros.3
Su inicio es insidioso, si bien a la larga puede ser súbito, con posibilidades de curarse de manera espontánea, desaparecer o permanecer inalterada durante meses o años. En términos clínicos se manifiesta como máculas purpúricas, que pueden confluir para formar placas de contornos irregulares con vasos puntiformes de aspecto de granos de pimienta de cayena, que no desaparecen a la vitropresión. Por lo general, son asintomáticas y simétricas, afectan los miembros inferiores en la región pretibial y pueden extenderse luego al resto de la piel, pero no afecta el rostro, las palmas y las plantas. A medida que la enfermedad evoluciona, adquiere tonalidad parduzca.3,4
El diagnóstico es principalmente clínico; se apoya en algunas técnicas para fundamentarlo, como la biopsia cutánea, cuyo estudio evidencia infiltrado perivascular linfohistiocitario localizado en los vasos de pequeño calibre en la dermis superficial. Es típico el edema en las células endoteliales y el estrechamiento de la luz vascular. También es frecuente observar la extravasación de hematíes con depósito de hemosiderina en los macrófagos.3
Se recomienda practicar una analítica sanguínea para descartar trombocitopenia, alteraciones en la coagulación o de la autoinmunidad (anticuerpos antinucleares, factor reumatoide) e infecciones crónicas (anti-VHC y anti-HBsAg).5
En el estudio dermatoscópico se evidencian glóbulos y puntos rojos que se deben a la extravasación de hematíes y al aumento de la cantidad y dilatación de los vasos sanguíneos; puntos marrones debido a la disposición esférica o elíptica de melanocitos y melanófagos en la unión dermoepidérmica sobre un fondo rojo-cobrizo difuso, por el infiltrado dérmico linfohistiocitario, la extravasación de hematíes y el depósito de hemosiderina en los histiocitos.3
No existe un tratamiento estandarizado; sin embargo, pueden aplicarse vía tópica esteroides de mediana y alta potencia, éstos reducen el prurito asociado y producen aclaramiento de las lesiones en algunas ocasiones.6
Debido a la cronicidad de las lesiones y a la necesidad de regímenes terapéuticos prolongados, los inhibidores de la calcineurina pueden considerarse una buena alternativa a los corticosteroides tópicos para conseguir aclaramiento o alivio de las lesiones de la dermatosis purpúrica pigmentaria.7 Asimismo, puede indicarse la fototerapia en los casos extensos o, bien, en los que no han respondido a corticosteroides o inhibidores de la calcineurina tópicos.8,9
Las opciones terapéuticas sistémicas incluyen la pentoxifilina como monoterapia, a dosis de 400 mg dos o tres veces al día durante dos a tres meses, o asociada con otros fármacos, como prostaciclinas (PGI2) y corticosteroides orales con muy buenos resultados.9
En cuanto a su pronóstico, a pesar de que las dermatosis purpúricas pigmentarias son dermatosis benignas y de alivio espontáneo, algunos casos se asocian con evolución a micosis fungoide, por lo que el diagnóstico temprano y seguimiento periódico anual son decisivos.8,9
Por último, debe tenerse en cuenta que las púrpuras pigmentarias son afecciones benignas, que constituyen un grupo de enfermedades crónicas con diferentes formas de manifestación clínica, de etiopatogenia poco conocida pero, aparentemente, con aspectos en común entre sí. Aunque las dermatosis purpúricas pigmentarias constituyen un grupo de enfermedades benignas, por lo general de alivio espontáneo, deben tomarse en cuenta por su heterogeneidad clínica, por los síntomas que llegan a ocasionar y porque, en algunos casos, se consideran formas asociadas con neoplasias linfoproliferativas.9
REFERENCIAS
1. Bonnet U, Selle C, Isbruch K, Isbruch K. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep 2016; 10 (1): 301. https://doi.org.10.1186/s13256-016-1065-6
2. Díaz V, Tirado A, Ponce RM. Dermatosis purpúricas y pigmentarias. Revisión. Dermatol CMQ 2009; 7 (3): 171-180.
3. Pallás IM, Conejero R, Biosca VL. Dermatosis purpúricas pigmentadas. Revisión de la literatura científica. Actas Dermosifiliogr 2020; 111 (3): 196-204. https://doi.org.10.1016/j.ad.2019.02.013
4. Her S, Amato V, Manzur G, Mássimo JA. Dermatosis purpuricas pigmentarias: a proposito de 5 casos en adolescentes. Dermatol Argent 2011; 17 (1): 26-31.
5. Çaytemel C, Baykut B, Ağırgöl Ş, Caf N, et al. Pigmented purpuric dermatosis: Ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol 2021; 48 (5): 611-616. https://doi.org.10.1111/cup.13949
6. Hilerowicz Y, Sprecher E, Gat A, Artzi O. Successful treatment of Schamberg’s disease with fractional non-ablative 1540 nm erbium: Glass laser. J Cosmet Laser Ther 2018; 20 (5): 265-268. https://doi.org.10.1080/14764172.2017.1418513
7. Jayakumar KL, Samimi SS. Jay Frank Schamberg, MD-beyond the eponymous disease. JAMA Dermatol 2017; 153 (12): 1242. https://doi.org.10.1001/jamadermatol.2017.4273
8. Errichetti E. Dermoscopy of inflammatory dermatoses (inflammoscopy): An up-to-date overview. Dermatol Pract Concept 2019; 9 (3): 169-180. https://doi.org.10.5826/dpc.0903a01
9. Harrison L. Dermatosis purpúricas pigmentarias. Revisión de tema. DER-RNT-7 2017; 1.
Este artículo debe citarse como: Bermúdez N, Dorta M, Vivas S. Enfermedad de Schamberg: caso inusual. Dermatol Rev Mex 2025; 69 (2): 309-312.
